
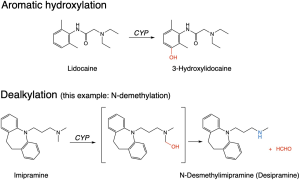
Cytochrome P450 (CYP) enzymes
A large family of enzymes found throughout the body, but concentrated in the liver, responsible for the majority of phase I metabolism of drugs. These enzymes take an oxygen atom from O2 and introduce it into a drug structure in a functionalisation reaction, so-called because this either adds or exposes a functional group. The functional group may then be targeted by a conjugating enzyme, or the phase I metabolite may already be sufficiently water-soluble to be eliminated in the urine.
There are several CYP enzymes important in drug metabolism; the two most important, based on the number of different pharmaceuticals that are substrates, are CYP3A4 and CYP2D6; together, these enzymes are involved in metabolism of the majority of administered pharmaceuticals.
CYP activity in the liver plays a large part in determining oral bioavailability of drugs, and in determining hepatic clearance of drugs. If a drug is a good substrate for one or more CYP enzymes (i.e. the drug is oxidised rapidly with a high kcat and a low KM) then that drug would be referred to as a high hepatic extraction ratio drug. Conversely, poor substrates that are oxidised much more slowly are referred to as low hepatic extraction ratio drugs.
Some CYP enzymes are products of polymorphic genes, meaning genes can exist in two or more forms, variant alleles differing in at least one position as a result of a nucleotide mutation or deletion. Most often, a mutation may have little effect or may reduce enzyme activity, while a nucleotide deletion is more likely to code for a protein that has lost activity. As a result, within a population, individuals with wild-type enzyme or with a mutation that has little impact on activity are referred to as extensive metabolisers or rapid metabolisers, while those with reduced activity are referred to as poor metabolisers. The frequency of poor metabolisers varies between individuals from different ethnic backgrounds. For example, up to 10% of Caucasians are poor metabolisers with respect to CYP2D6, whereas only 1% of individuals of Asian descent are CYP2D6 poor metabolisers. The pattern is reversed for CYP2C19, with around 4% of Caucasians and 16% of Asians possessing a poor metaboliser phenotype.
CYP enzymes vary significantly in their expression levels between individuals and cause much of the variability in drug responses observed between different individuals. The consequences of this variation are exacerbated by changes in CYP activities that occur as individuals age (CYP activity is lower in the elderly population) and in several disease conditions – particularly hepatic pathologies. Further, numerous drugs, environmental chemicals (such as those present in cigarette smoke or pesticides), and dietary components found in barbecued meat and in cruciferous vegetables, can induce expression of a number of CYP enzymes, thereby modifying first pass metabolism and/or hepatic clearance of drugs that are substrates for the enzymes affected. Compounds from the furanocoumarin family that are present in grapefruit juice act as irreversible inhibitors of CYP3A4, potentially elevating oral bioavailability and/or reducing clearance of CYP3A4 substrates.
As a consequence of enzyme induction, irreversible enzyme inhibition and competition between two or more drugs for binding to the enzymes’ active sites, CYP enzymes are also major contributors to the prevalence of drug-drug interactions.
CYP activity versus a particular drug is often the most important contributor to the value for intrinsic clearance (Clint) of that drug.
Clinical Context
From a clinical perspective, CYP enzymes are at the root of many drug interactions. Medications may compete for binding to one or more CYP enzymes, and/or they may induce expression of these enzymes. Co-administration of two medications that are substrates for the same CYP enzyme(s) is a common cause of “drug-drug interactions”, as a result of competition and increased levels of both drugs. If one of the medications inhibits CYP metabolism of the second medication, this can lead to a relative “overdose” of the latter medication. For example, a bone marrow transplant patient using the anti-rejection medication tacrolimus may develop an invasive fungal infection and require initiation of fluconazole. Tacrolimus and fluconazole are both substrates for CYP3A4. Fluconazole inhibits metabolism of tacrolimus, which has a narrow therapeutic window, potentially leading to the accumulation of toxic levels of tacrolimus. It is common practice to reduce the dosing rate of tacrolimus by ⅔ when initiating fluconazole treatment, in anticipation of the increase in levels of tacrolimus that occur as a result of CYP3A4 inhibition.
Medications may also upregulate production of CYP enzymes and induce their own metabolism, or the metabolism of co-administered medications. Examples of commonly-used medications that induce CYP expression are the antiepileptic phenytoin and the antibiotic rifampin.
Management of these issues can be quite straightforward if the affected medication remains at therapeutic levels within the therapeutic window. However, this isn’t the case with most medications. Clinicians need to balance the risk of potentially causing toxicity from excessive exposure due to decreased metabolism with the risk of treatment failure due to under-dosing when co-administering two medications. There is rarely direct evidence to support this decision-making process, but clinicians may use a combination of published pharmacokinetic data, or case report data, in combination with careful monitoring of drug levels in the blood, to try to rationalise decision-making.
Certain classes of medications are well-known to have the potential to cause problems through CYP interactions. One should always be aware of potential interactions, but medication classes including antiepileptics, antifungals, anticancer drugs, antidepressants, antipsychotics and transplant medications are well-known for this issue.
