
Apparent volume of distribution
The definition of the apparent volume of distribution (AVD) is “the apparent volume of plasma that would contain the total body content of the drug at a concentration equal to that in the plasma”. As such, AVD (sometimes referred to simply as the volume of distribution, VD) is a theoretical volume that provides an indication of the extent to which a drug distributes from the bloodstream into tissues and organs. In essence, the AVD value represents an indirect means of expressing the proportion or ratio of an administered drug dose that remains in the plasma at equilibrium, with AVD inversely-related to the percentage of a dose that remains in the plasma at equilibrium. Calculated volumes may bear no relation to physiological volumes associated with part or all of the body, and may be very much larger than a patient’s total body volume. However, by expressing extent of distribution as a volume term, rather than in a manner that is based on drug concentration, the approach remains independent of dose, for most drugs, and the value for AVD is typically independent of the dose of drug administered. It is necessary to know a drug’s AVD in order to calculate the amount of drug present in the body that remains to be excreted, or to determine a loading dose.
Click here to view a short vodcast offering a simple, largely qualitative explanation of how AVD is determined and how AVD varies with increasing extent of distribution.
One-compartment drugs
The AVD is nominally obtained by dividing the dose of a drug administered by the theoretical concentration in the plasma after distribution is complete, but before any drug has been eliminated. In other words, all the drug is still in the body, and it has reached an equilibrium between the blood and the tissues. For a one-compartment drug, this theoretical concentration (referred to as Cp0, or concentration in the plasma at time = 0) is obtained from the intercept with the Y-axis (concentration axis) of a plot of drug concentration in the plasma versus time. The intercept is obtained by back-extrapolating the fitted curve to the Y-axis, a process facilitated by plotting data on a logarithmic scale so that data resulting from the single exponential process (elimination) appear linear.
As a result of rapid drug movement between the plasma and tissues, the percentage of drug in the body that is located in the plasma remains constant from the point when equilibrium is reached until all drug has been cleared from the body, regardless of how rapidly drug is administered into, or is cleared from, the bloodstream. In other words, the AVD remains constant. The initial volume of distribution is equal to the central compartment volume, and is also equal to the terminal volume of distribution. The following equations are both acceptable for calculating AVD for a one-compartment drug, and would be expected to yield similar or identical values.
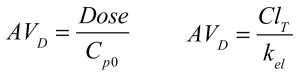
Two-compartment (or multi-compartment) drugs
The equivalent process to calculate AVD for a two-compartment drug would use the B-intercept of a hockey-stick plot, in place of Cp0. However, determining AVD for a two-compartment drug in this manner results in calculation of what is referred to as an extrapolated AVD (AVD extrap), which often over-estimates AVD (in that multiplying the concentration of drug in the plasma by AVD could significantly over-estimate the amount of drug present in the body). In fact, as a result of the slower movement of a two-compartment (or multi-compartment) drug between plasma and tissues, AVD can have different values, and subtly-different meanings, depending upon the point in time at which AVD is determined. As such, there are several approaches to calculating AVD for a two- (or multi-) compartment drug, with AVD extrap representing the least-useful of these. Of more value, clinically, are the non-compartmental volume of distribution (AVD area, also referred to as AVD beta) and the volume of distribution at steady state (AVD SS).
Click here to view a vodcast demonstration of the differences between AVD area and AVD SS.
If a drug is infused into plasma until a steady-state concentration is reached, there is neither net movement of drug between plasma and tissues nor net movement of drug into or out of the body. At that point, the ratio of drug concentration in the tissues to drug concentration in the plasma would have reached a constant value, and dividing total drug in the body by drug concentration in the plasma would yield a value for AVD SS. If drug infusion is then terminated, the concentration of drug in the plasma will initially fall more rapidly than that in the tissues. The lag in redistribution of drug from tissues back to plasma persists such that a new position of pseudo-equilibrium is reached, with a new, higher ratio of drug concentration in the tissues to drug concentration in the plasma. This ratio again remains constant as drug concentrations fall in both compartments. At any point after pseudo-equilibrium is reached, dividing total drug in the body by drug concentration in the plasma would yield a value for AVD area, which is typically a little larger than the AVD SS.
If total body clearance for a drug is relatively low, the lag caused by redistribution of drug from tissues to plasma is small, and the AVD area is similar in magnitude to the AVD SS. However, high clearance rates, simulated in hydraulic models by increasing the flow rate of the tap flushing fresh water through the central compartment, can cause a marked increase in the ratio of drug concentration in the tissues to drug concentration in the plasma, and under those circumstances, AVD area can be markedly higher than the AVD SS. The larger impact of clearance on AVD area explains the larger reduction in AVD area that occurs in renal insufficiency, compared with the more modest effects on AVD SS.
Determination of AVD area can be achieved through applying either of the equations below to patient data, and is quite straightforward.
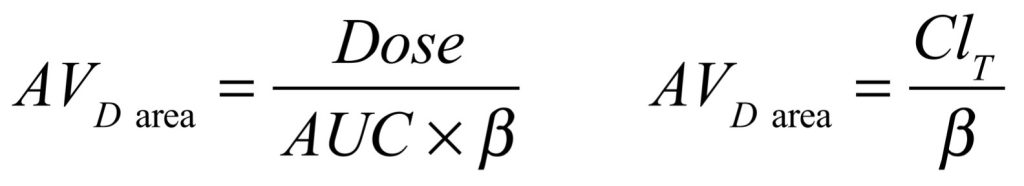
Since the AVD area is determined under pseudo-equilibrium conditions where drug elimination from the plasma and slow redistribution from the tissues lead to a lag that increases the tissue:plasma ratio of drug, the AVD area is the correct value to use to address clearance-related questions such as that of how much drug remains to be eliminated from the body. In contrast, the AVD SS is determined, by definition, when infusion and clearance rates are identical, and there is no net clearance of drug. As such, the AVD SS is the value that should be used to calculate a loading dose (if administered orally, bioavailability must be accounted for, if less than 100%):
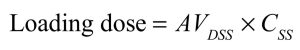
In this scenario, a bolus dose (or a series of bolus doses, or a brief infusion) equal in total to the entire amount of drug that would be present in the body at steady state with repeated dosing is administered over a relatively short time period prior to commencing a maintenance dosing regimen, in order to achieve an effective therapeutic concentration of drug as quickly as possible. This may be useful when administering an antibiotic to treat a severe or life-threatening infection.
Determining a value for AVD SS from patient data is rather less straightforward than for other forms of AVD, and is not discussed further here. Nevertheless, students should be familiar with the term, and its meaning, as this is arguably the most clinically-useful form of AVD. In this regard, many literature sources will provide values specifically for AVD SS.
General comments
The more extensively the drug distributes to tissues, the lower the remaining concentration of drug in the plasma, and so the larger the (apparent) volume in which the entire dose of drug appears to have been dissolved. Therefore, a large AVD (e.g. 500 litres, and potentially several thousand litres) indicates that the drug has accumulated extensively in the tissues (and is therefore probably a very lipophilic drug), while a value of around 10 litres suggests that most of the drug has remained in the plasma – probably as a result of high plasma protein binding. A very low volume of distribution can also be observed for a charged or extremely polar drug that is administered by IV injection or infusion and that is unable to distribute to an appreciable extent from the plasma because of its polarity. An example of a drug class that behaves in this way would be aminoglycoside antibiotics. Due to its low lipophilicity, an IV dose of an aminoglycoside remains largely in the plasma and is eliminated rapidly and extensively by the kidneys, making these drugs ideal for treating serious urinary tract infections. In contrast, an oral dose is poorly absorbed from the GI tract, and a PO dosing regimen is thus effective for “sterilising” the GI tract prior to GI surgery.
Though an AVD of around 35 litres may appear to suggest that most of the dose has distributed throughout body water, such an observation would rather indicate that similar proportions of drug remain unbound (to plasma proteins) in the blood, and unbound (to tissue proteins) or unsequestered (by fat), in the tissues. When a calculated AVD has a volume similar to that of a physical body compartment, this is often coincidental.
The factors that contribute to the numerical value for AVD can be better understood if the form of an equation that contains these factors is considered.
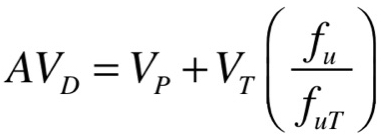
VP and VT refer, respectively, to the volume of the plasma (approximately 4 litres) and the volume of total body water minus the plasma volume (approximately 30-35 litres). The factor in parentheses contains terms for fu, the fraction of drug unbound in the plasma, and fuT, the fraction of drug unbound in the tissues. “Bound” can be loosely defined to mean that the drug is not immediately available in an aqueous environment to interact with target proteins, enzymes or transporters, or to diffuse through cell membranes. As such, drug bound in the plasma would typically be bound to soluble plasma proteins, although binding to other components of the cell wall is also possible, while drug bound in the tissues could be bound to proteins (target proteins or non-specific binding to other proteins) or is unavailable as a result of significant partitioning into fat.
The equation can’t be used to determine AVD in a patient, because it is not feasible to determine a value for the fuT term. Rather, it serves simply to assist in understanding the factors contributing to AVD.
It should be apparent that the value for AVD is equal to the plasma volume (4 litres), plus the total body water volume (30-35 litres) multiplied by the factor in parentheses. Thus, if fu is small compared with fuT (in other words, drug is highly bound to plasma proteins but is not highly bound to proteins in tissue and also has not partitioned extensively into fat) then the term in parentheses will have a value lower than 1 and AVD will lie between 4 litres and 34-39 litres. Alternatively, if the drug has partitioned very extensively into fat such that fuT is much smaller than fu, even if the drug is highly bound to plasma proteins (for example, fu is 0.05 and fuT is 0.002) then the term in parentheses is a value much higher than 1 (in this example, 25) and so AVD would equal 4 litres + (30 litres × 25), or around 754 litres. This illustrates why a high level of plasma protein binding does not mean AVD must be small. In this regard, it is necessary to know both a drug’s AVD and the degree to which it binds to plasma proteins in order to form a fuller understanding of the drug’s disposition in the body.
The terms present in this equation demonstrate the importance of a patient’s plasma protein levels, and of their body fat content, to their personalised value for AVD for a particular drug. It is also important that plasma volume and total body water volume can impact the value for AVD quite markedly. Since these volumes can change substantially in clinical conditions such as septic shock, or following IV fluid infusion to address hypotension, or with renal and cardiac pathologies that lead to oedema, then AVD for drugs with a small volume of distribution can be altered accordingly, often leading to inadequate or unexpectedly high drug concentrations in the plasma. In contrast, the effect on AVD for lipophilic drugs is small, since the vast majority of drug in the body exists outside the plasma, and even a significant change of the concentration of unbound drug in the plasma would be quickly nullified by drug moving between plasma and adipose tissue.
Click here to view a short vodcast on drug properties impacting the numerical value of AVD.
Clinical Context
Changes in volume of distribution can be important considerations in dosing of medications and monitoring treatment outcomes. Understanding whether a medication distributes from the circulation predominantly into the extracellular fluid, into fatty tissue, or whether it distributes into both may be helpful in dosing and monitoring your patients. With antimicrobial medications, for which drug concentrations at the site of infection can be critically important, fluid shifts can be an important factor to consider in monitoring patient improvement. Compare, for example, an obese, fluid-overloaded congestive heart failure (CHF) patient with a non-fluid-overloaded non-obese patient, both treated with vancomycin to treat a severe infection such as septic arthritis caused by methicillin-resistant S. aureus (MRSA). The obese, fluid-overloaded patient would likely require loading doses to reach effective drug concentrations quickly, and would require higher doses to achieve target levels. As vancomycin distributes extensively to extracellular fluid, but far less extensively to body fat, the fluid-overloaded patient would require a higher dose of vancomycin per kilogram of bodyweight to maintain effective target trough levels where they are measured – in the blood. It is important that vancomycin concentrations in the blood are maintained above a minimum effective concentration for the duration of the treatment period. Vancomycin has the advantage of having serum levels readily available to monitor this patient.
